9. Molecules
A molecule is an assembly of atoms held together in a stable conformation by chemical bonds. A chemical bond, in turn, is a reasonably stable connection between atoms (this definition is by Roald Hoffmann, chemistry Nobel laureate 1981). Molecules exist because their energy is lower compared to the separated constituent atoms. In this chapter, we lay out molecular orbital (MO) theory, which allows us to predict the energy and the geometric and electronic structure of molecules. Recall that a molecular orbital (MO) is a one-electron wavefunction in a molecule.
For the purpose of quantum theoretical description, a molecule is considered a collection of nuclei and electrons. The total molecular Hamiltonian for such a collection of particles is
The first and second sum collect all kinetic-energy terms, for all electrons
Note
For H2O, there are 10 electrons and 3 nuclei, so there are 13 kinetic-energy terms, 30 attraction terms, 3 nuclear repulsion terms, and 45 electron-electron repulsion terms. This gives a total of 91 terms.
With such a Hamiltonian, the Schrödinger equation is unsolvable, even when using the orbital approximation (i.e. a Slater determinant form of the wavefunction). To simplify the problem, another important approximation is made. We discuss this next.
9.1. Born-Oppenheimer approximation
The Born-Oppenheimer approximation is a notion of central importance to chemistry. The reasoning for it goes as follows. Electrons and nuclei experience similar electrostatic forces in a molecule. Now, since nuclei are much more massive than electrons (e.g. the proton mass is 1836 times that of the electron), nuclei are accelerated much less by these forces. Therefore, at very short timescales (less than 10-16 seconds), electrons move, but nuclei essentially don’t. From the point of view of the electrons, the nuclei appear frozen; and from the point of view of the nuclei, the electrons appear not as localized particles, but as a continuous delocalized distribution of negative charge.
Note
As a useful visual analogy for this, picture a nucleus as a hiker, and the surrounding electrons as mosquitoes. The mosquitoes move much faster than the hiker (unless she runs), therefore the hiker perceives them as “delocalized”, whereas the mosquitoes see the hiker as essentially stationary.
This separation of time scales allows us to separate nuclear and electron motion. This is the Born-Oppenheimer approximation. The total wavefunction
Here,
where
Now a two-step solution is possible. First, the nuclei are assumed fixed at specific positions, and the Schrödinger equation for the electrons is solved, yielding the electronic wavefunction
9.2. Dihydrogen cation
The simplest molecule is the dihydrogen cation,
The electronic Hamiltonian for
Figure 9.1 gives the meaning of the various distance variables,
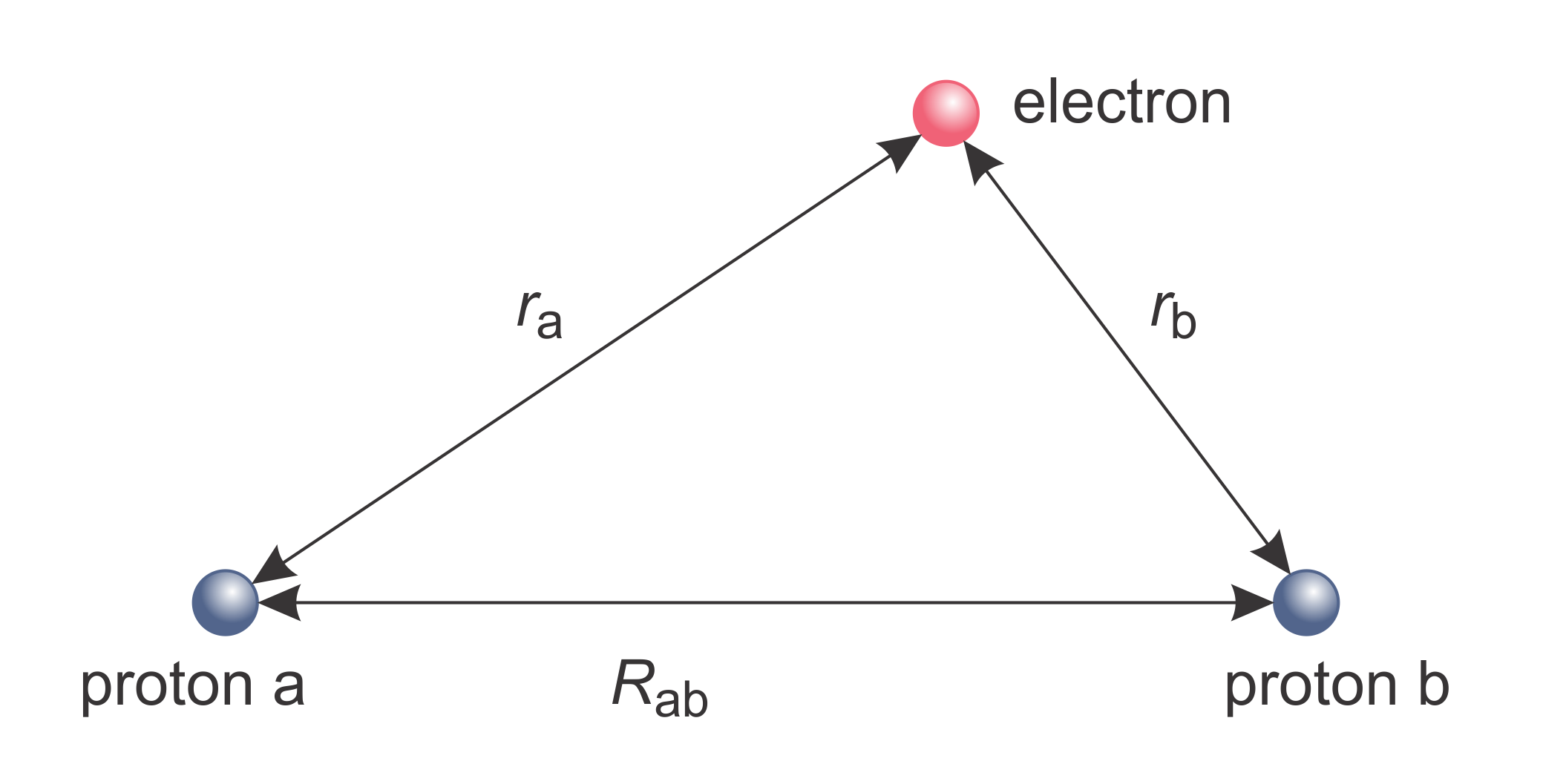
Figure 9.1 Dihydrogen cation
Energies. Solving the electronic time-independent Schrödinger equation for a range of
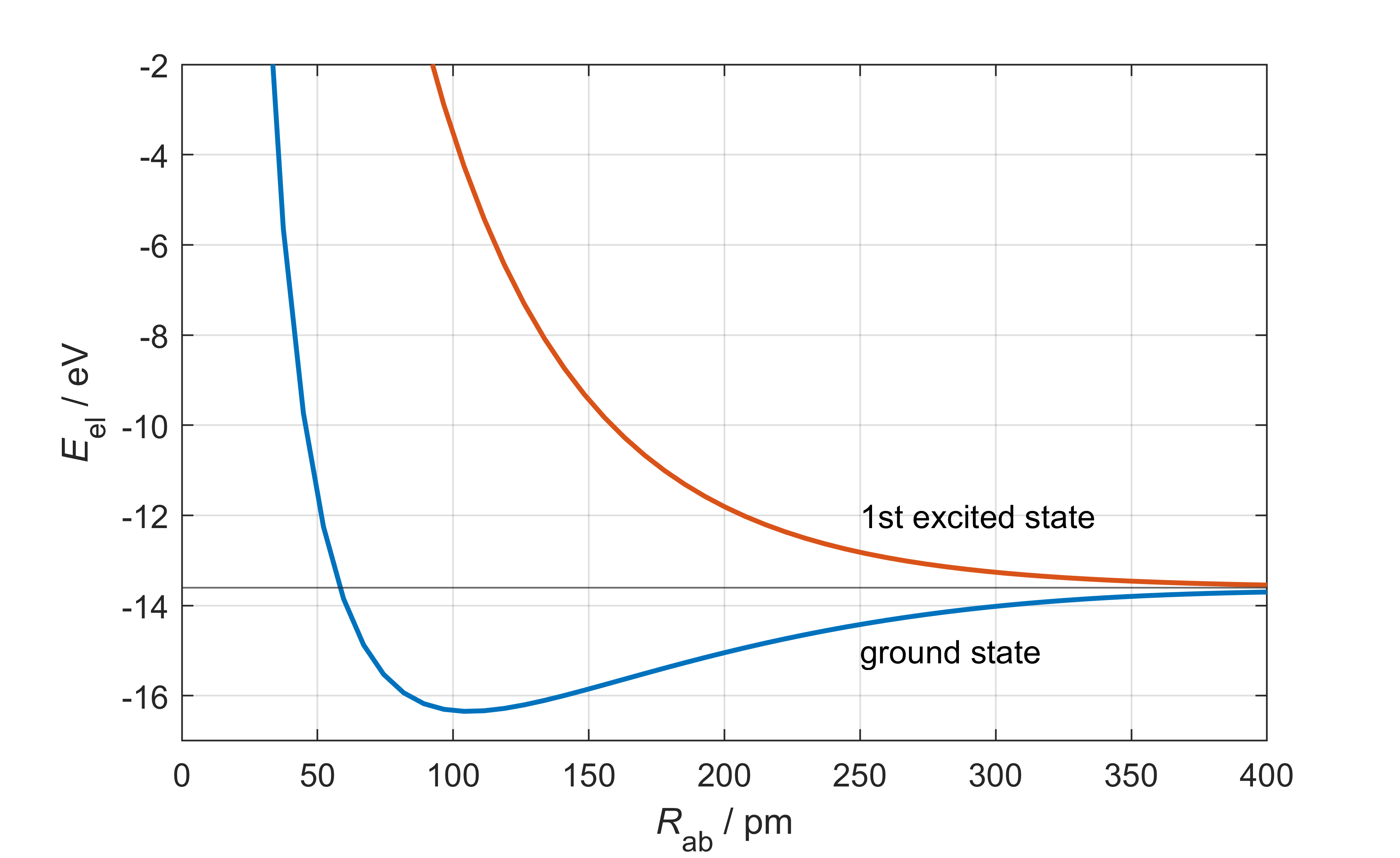
Figure 9.2 The potential-energy curves of the electronic ground state (blue) and first excited state (red) of
The ground-state energy shows a minimum at about 105.7 pm, indicating that two protons and an electron form their lowest-energy state at this distance. At the minimum, the energy is -16.3 eV, about 2.7 eV lower than at infinite separation (
Wavefunctions. The wavefunction for the electronic ground state at the equilibrium geometry (105.7 pm) is shown in Figure 9.3. Near the nuclei, the wavefunction resembles closely the AOs of the constituent atoms. In addition, there is increased electron density between the two nuclei, such that the wavefunction is more delocalized than the AOs of each constituent atom.
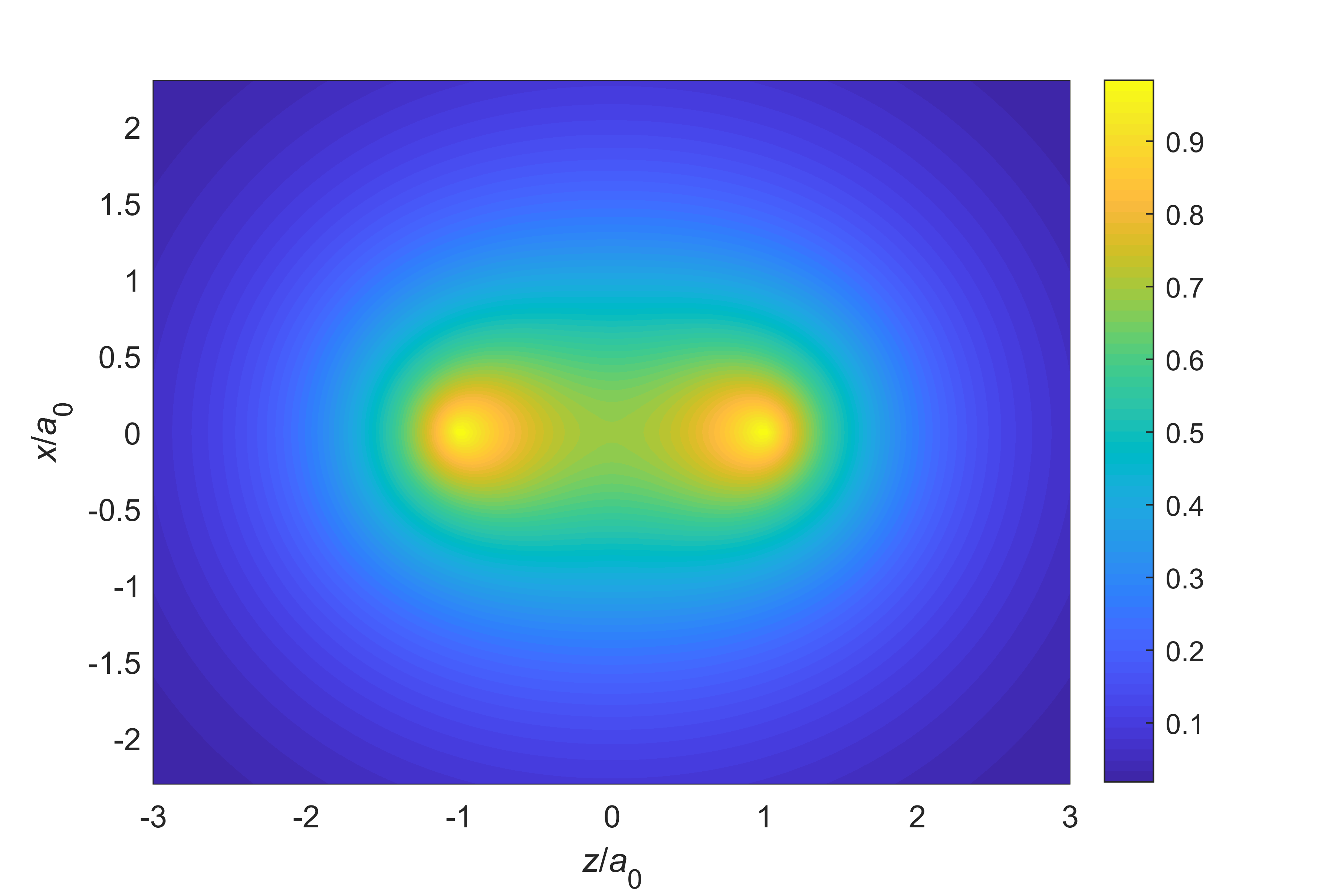
Figure 9.3 The ground-state wavefunction for the dihydrogen cation, for the equilibrium inter-nuclear distance 105.7 pm (= 2
Note
Why is this molecule stable? There are two reasons: (a) The electron has more space to spread out in
A purely electrostatic explanation for the stability, in terms of increased electron density between the two nuclei, is often taught in introductury chemistry courses. It is incomplete, as is misses to take into account the electron motion. It also suggests incorrectly that the location of the lowest potential energy for the electrons is between the nuclei - that is actually close to the nuclei.
Since this is a one-electron molecule, this is a one-electron wavefunction, i.e. a molecular orbital. For molecules larger than
9.3. MO theory
Now, the molecular orbitals of the molecular hydrogen cation shown in Figure 9.3 look very similar to the 1s atomic orbitals near the nuclei. This suggests that a reasonable strategy for simplifying the problem and developing a general strategy for calculating MOs would be to approximate the MOs as linear combinations of AOs or basis functions that resemble AOs (LCAO). This method is called MO-LCAO and is the cornerstone of MO theory.
As a minimal example, consider the following approximate MO built from two AOs or AO-like basis functions
where
with the abbreviations
The linear-combination coefficients
Each of these equations results in a linear equation in
Solving this quadratic equation gives
For a homodiatomic molecule (
Since the MO contains two AOs, we have two linear-combination coeffients (
Figure 9.4 shows the accompanying orbital energy level diagram, called the MO diagram, with the case of hydrogen indicated, where
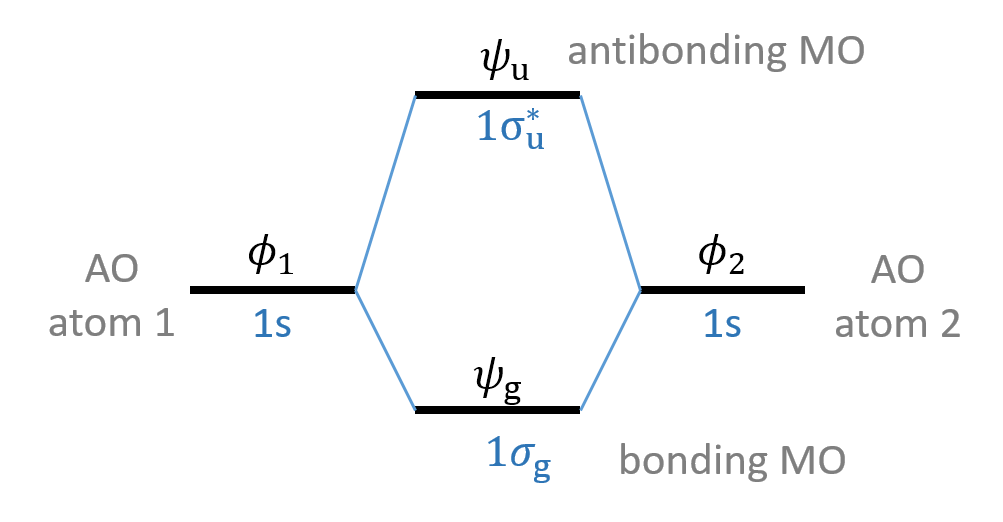
Figure 9.4 MO diagram involving two atomic orbitals.
The MOs built from two 1s functions are shown in Figure 9.5. The sum of the two 1s functions gives the bonding MO, with increased electron delocalization. The difference gives the antibonding MO, with reduced electron delocalization.
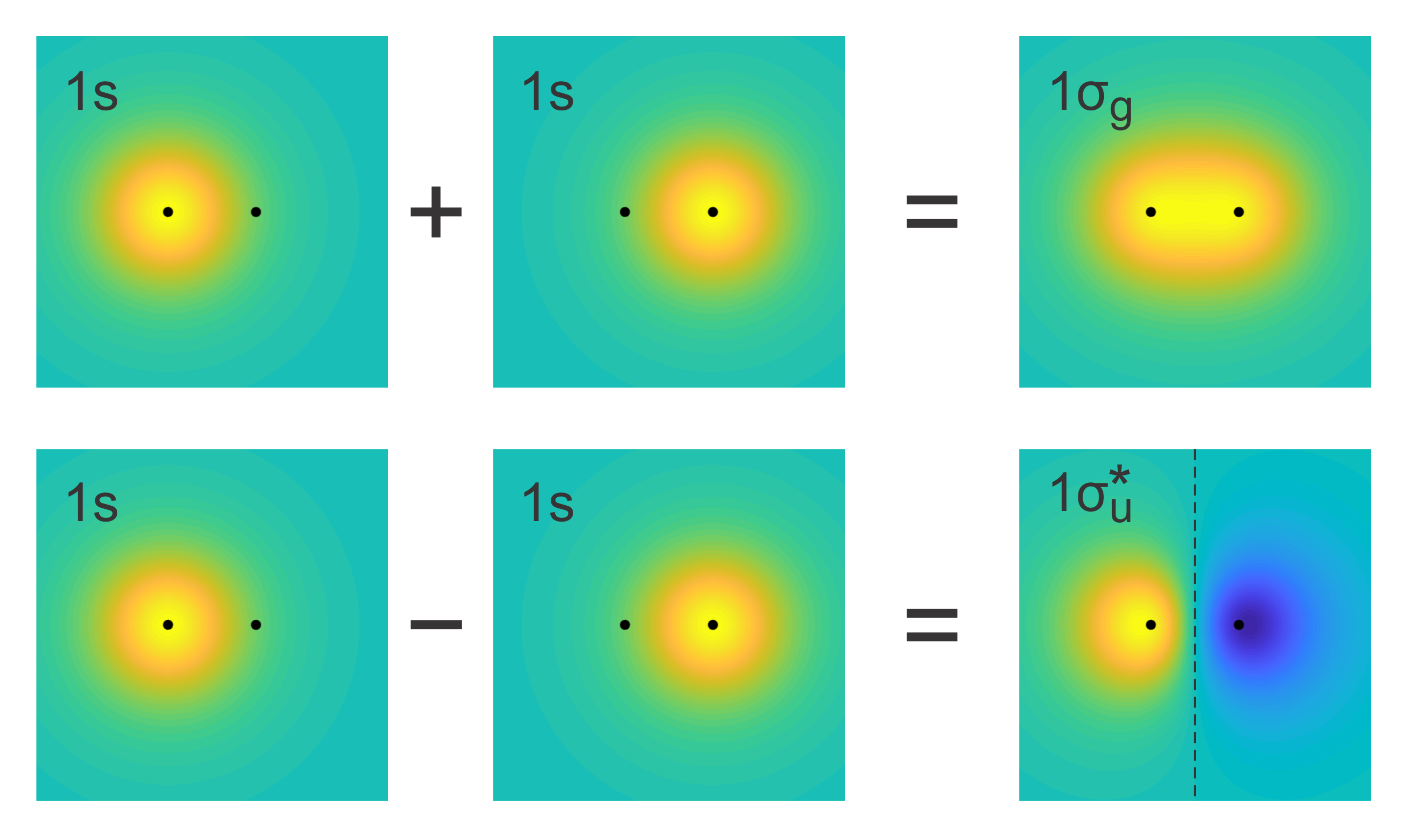
Figure 9.5 Linear combinations of two 1s atomic orbitals give a bonding MO and an antibonding MO. The black dots indicate the locations of the nuclei, and the dashed line indicates the nodal plane perpendicular to the bond. Yellow indicates positive, blue negative, and green close-to-zero values.
Figure 9.6 summarizes the nomenclature for MOs in diatomic molecules. It abbreviates information about the symmetry and the energy of the MO. The Greek letter indicates the symmetry of the MO around the bond axis:
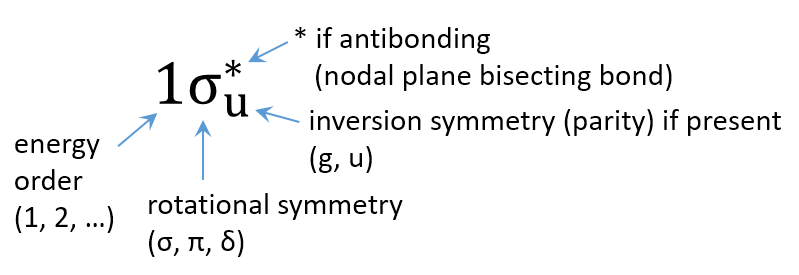
Figure 9.6 Labels for orbitals in diatomic molecules.
Note
For example,
Given the MOs, the electron configuration for a given number of electrons can now be determined using the Aufbau principle (as discussed for atoms): Fill the
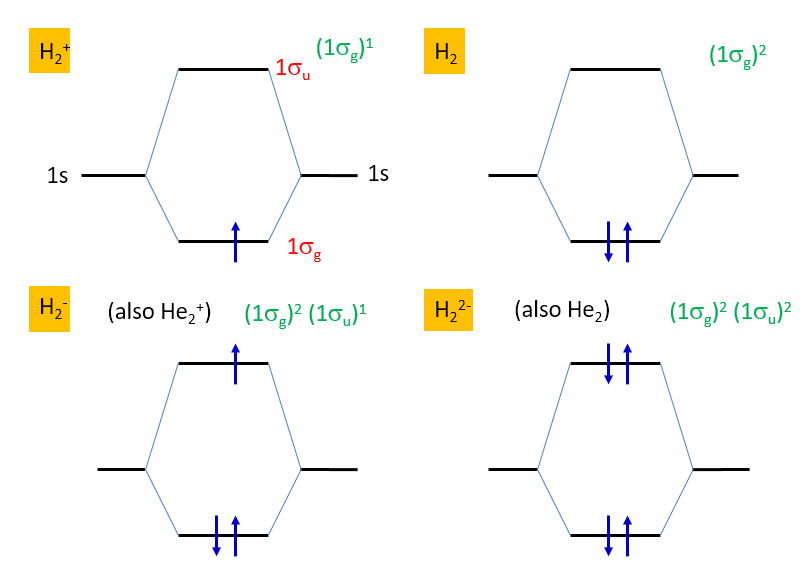
Figure 9.7 MO diagrams illustrating the Aufbau principle for the various charge species of dihydrogen. Electron configurations are shown in green.
Second-row diatomics. For second-row elements, the additional AOs from the second shell determine the bonding and antibonding MOs. The 1s AOs are more concentrated around the nuclei and do not overlap. With the 2s and 2p orbitals, several types of
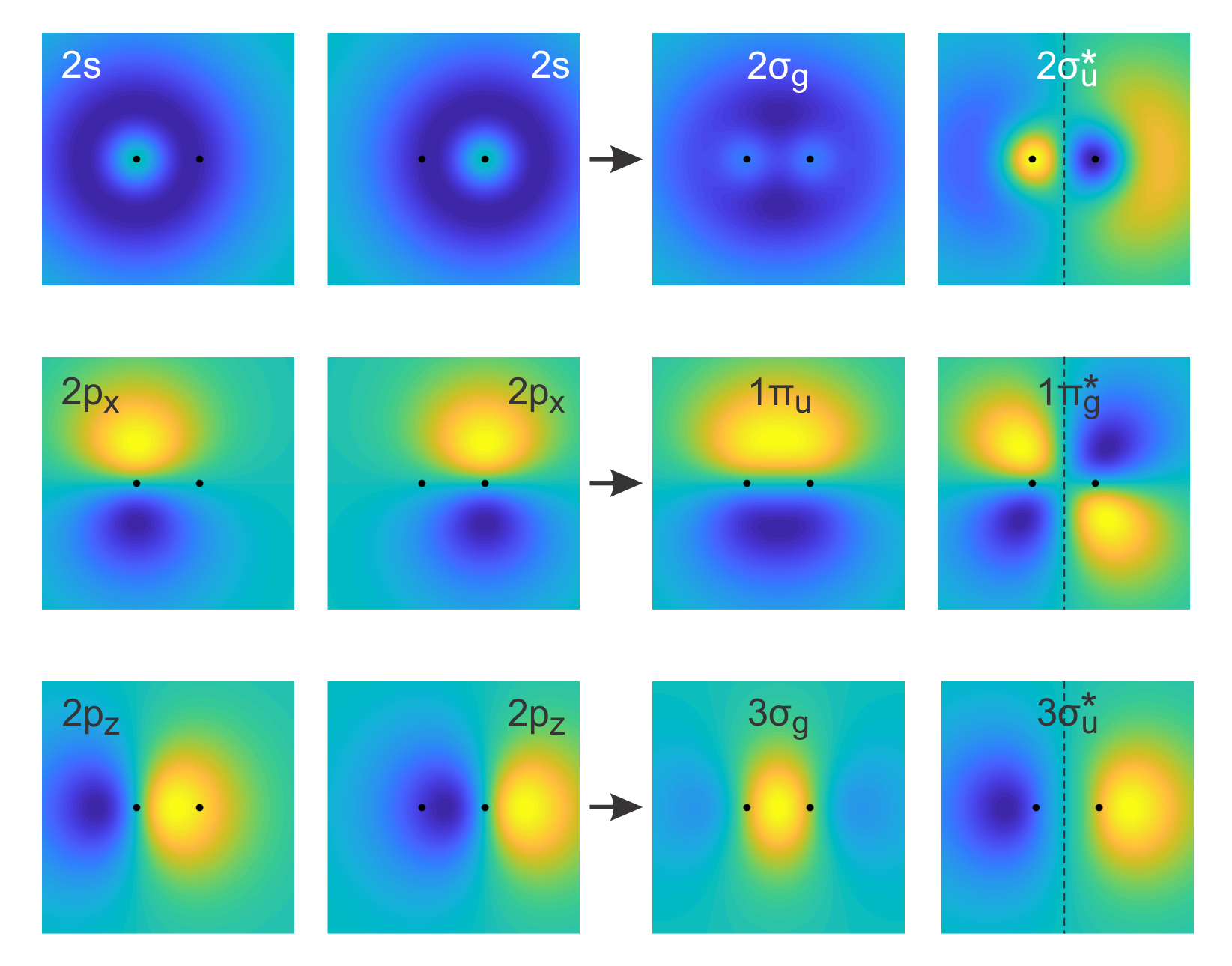
Figure 9.8 MOs contructed from linear combinations of AOs with
The progression of MO energies in neutral homodiatomic molecules across the second row of the periodic table are shown in Figure 9.9.
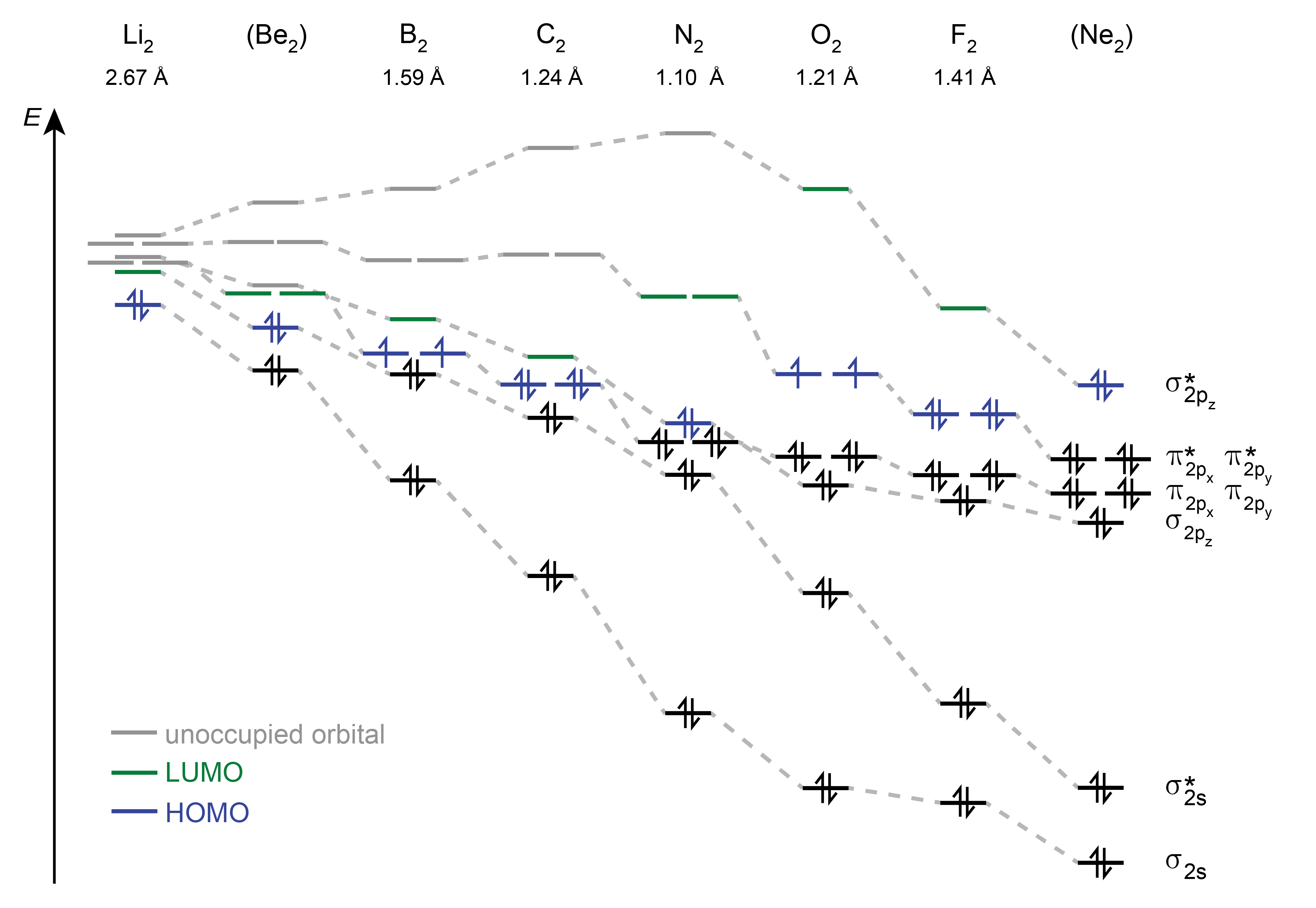
Figure 9.9 The MO diagrams of all homonuclear diatomics from Li to Ne. The two lowest-energy MOs resulting from the overlap of the 1s AOs are not shown. Note that Be2 and Ne2 have not been observed.
Koopmans’ theorem, as discussed in the previous chapter for atoms, applies to molecules as well. It states that the ionization energy is equal to the negative MO energy of the HOMO. This is an approximation, as removing one electron from a molecule does not just empty the HOMO, but also leads to a slight redistribution of the remaining electrons, and therefore to shape changes in the remaining occupied orbitals. This orbital relaxation is neglected in Koopmans’ theorem. This approximation is called the frozen-core approximation.
Bond order. The bond order in a diatomic molecule is defined as
If the total number of electrons is even, the bond order is an integer (0, 1, 2, 3). If it is odd, the bond order is a half-integer (1/2, 3/2). Figure 9.10 shows the bond orders for neutral homodiatomic molecules of the first two rows of the periodic table.
The bond order is a qualitative predictive tool. When comparing two molecules with different bond orders, the one with the higher bond order usually has (a) the higher bond dissociation energy, (b) the shorter bond length, and (c) the larger force constant.
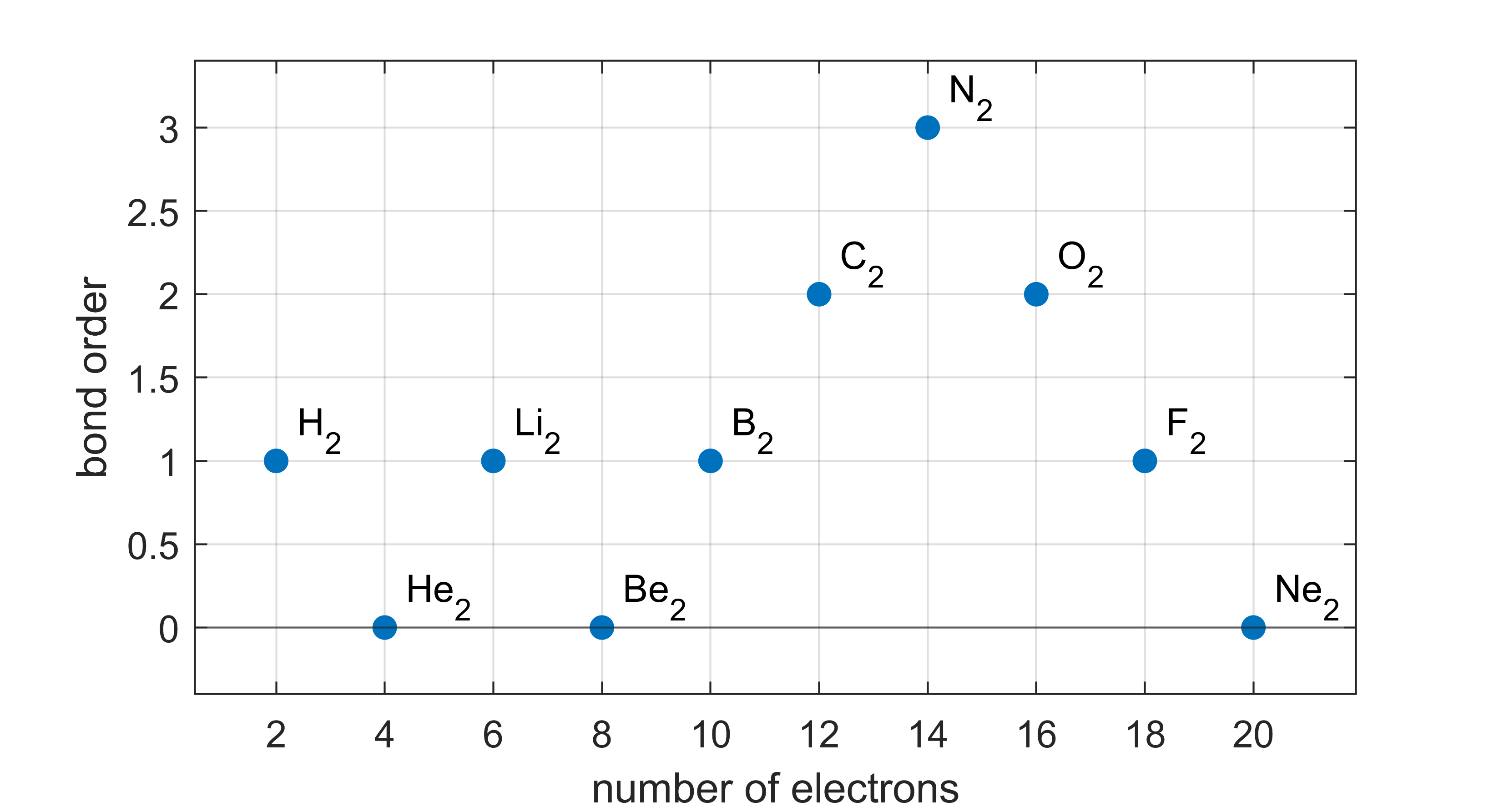
Figure 9.10 The bond orders of the homodiatomic molecules of the first and second row of the periodic table. Note that He2, Be2, and Ne2 have not been observed experimentally.