Page 1 of 2
What is a proper value of "Opt.Allocation"?
Posted: Sat Nov 19, 2016 1:59 am
by hirawk
I am studying molecular orientation and dynamics of nitronyl nitroxide radicals in confined spaces.
When I updated EasySpin from ver. 4.5.5 to 5.1.5,
the messages was displayed that "Diffpa" command should be changed to "DiffFrame",
and that the orders and signs of Eular angles should be converted.
However, although I following those orders and changed the document,
an error massage to increase the value of allocation were displayed.
I succeeded the calculation in ver. 4.5.5 by change "Opt.Allocation" to [1e7 2e5].
However, after the update to ver. 5.1.5, it was not good even when Opt.Allocation set to [4e7 2e5].
Please give me your advice.
Re: What is a proper value of "Opt.Allocation"?
Posted: Sat Nov 19, 2016 4:17 pm
by Stefan Stoll
Please post a minimal example that generates this error message.
Re: What is a proper value of "Opt.Allocation"?
Posted: Sat Nov 19, 2016 9:37 pm
by hirawk
Dear Stefan
I uploaded the following three files.
1. The one succeeded in 4.5.5
2. The one complained in 5.1.5
3. The original spectrum quoted in the above files.
Thank you for your kindness.
Re: What is a proper value of "Opt.Allocation"?
Posted: Sun Nov 20, 2016 3:50 pm
by Stefan Stoll
There were some memory allocation inefficiencies in version 5.1.5 that prevented your simulation from running cleanly. Thanks for identifying them.
We just posted version 5.1.6, where these problems are fixed. The following simulation, based on your code, now runs without problems:
Code: Select all
clear
Sys.g = [2.0118,2.0075,2.0031];
Sys.Nucs = '14N,14N';
Sys.A = mt2mhz([3.4,1.2,17.4;3.4,1.2,17.4]/10);
Sys.DiffFrame = [-78 -68 0]*pi/180;
Sys.Diff = [.01 .01 10]*4.05e6;
Sys.lw = [0.11, 0.21];
Exp.mwFreq = 9.064039;
Exp.CenterSweep = [323.2933 30];
Opt.Allocation = [4e7 2e5];
[B2,spc2] = chili(Sys,Exp,Opt);
plot(B2,spc2);
Re: What is a proper value of "Opt.Allocation"?
Posted: Sun Nov 20, 2016 6:30 pm
by hirawk
Thank you for your assistance.
I tried both of your attached code and my own with ver. 5.1.6.
However, the same error messages were presented as below.
error(in Japanese): chili_lm2
Number of non-zero elements too large. Increase Opt.Allocation(1).
error(in Japanese): chili
error(in Japanese): chili
Re: What is a proper value of "Opt.Allocation"?
Posted: Sun Nov 20, 2016 8:33 pm
by Stefan Stoll
That is surprising. Make sure you run EasySpin 5.1.6 by calling
and that you are using the right version of
chili by running
Re: What is a proper value of "Opt.Allocation"?
Posted: Mon Nov 21, 2016 12:22 am
by hirawk
Both versons of EasySpin and chili were 5.1.6 toghether.
Although I tried your codes and mine many times with changing parameters in one way or another,
the calculations were always terminated.
In particular, when maxElements of Opt.Allocation was set more than 3.1 e7,
my conputer was terminated.
Surrounding
PC: Panasonic CF-S10; Win7; Intel(R) Core(TM) i5-2540M CPU @ 2.60 GHz 2.60 GHz; RAM 4.00GB; 64 bit OS
MATLAB 2016b
True, my codes were running in ver. 4.5.5...
Re: What is a proper value of "Opt.Allocation"?
Posted: Mon Nov 21, 2016 2:42 pm
by Stefan Stoll
The Liouvillian for this problem has 30903072 non-zero elements, so the first element in
Opt.Allocation has to be equal or larger than that number.
Here is the output I get when running the example from above with
Opt.Verbosity = 1:
Code: Select all
-- slow motion regime simulation ----------------------------------
field sweep, mw frequency 9.064039 GHz
field range (mT): min 308.293, max 338.293, center 323.293, width 30
harmonic 1, perpendicular mode
No ordering potential given, skipping powder simulation.
Solver: Lanczos tridiagonalization, left-to-right continued fraction evaluation
using S=1/2 Liouvillian code
Ordering potential: absent
Setting up basis set...
spatial basis: Leven max 14, Lodd max 7, Kmax 6, Mmax 2, deltaK 1, jKmin -1
spin basis: pSmin +0, pImax 2 spin basis: pSmin +2, pImax
M-p symmetry: 0
basis size: 136080
Computing starting vector...
vector size: 136080x1
non-zero elements: 9/136080 (0.01%)
maxabs 0.333333, norm 1
Computing Liouvillian matrix...
size: 136080x136080, maxabs: 1
non-zero elements: 30903072/18517766400 (0.17%)
Computing spectrum...
converged to within 1e-06 at iteration 180/136080
Convoluting with Gaussian (FWHM 0.11 mT)...
-------------------------------------------------------------------
Re: What is a proper value of "Opt.Allocation"?
Posted: Mon Nov 21, 2016 6:46 pm
by hirawk
Thank you for your reply.
1. Even if I used the same calculation with another PC or MATLAB surroundings,
it was impossible so far.
I search a PC surrounding making it possible to achieve this calculation.
2. In the calcullation of a similar but easier system of mono-nitroxide radicals,
I tried the same calculation.
(This was also possible in ver. 4.5.5.)
The caluculation itself was finished.
However, all values became zero.
I also attached the files about 2.
Please check them again.
Re: What is a proper value of "Opt.Allocation"?
Posted: Mon Nov 21, 2016 6:57 pm
by Stefan Stoll
Based on your code, I ran the following example:
Code: Select all
clear all, close all, clc
Sys.g = [2.0095,2.0066,2.0023];
Sys.Nucs = '14N';
Sys.A = mt2mhz([5.9,4.8,33.3]/10);
a = 57.1e6;
Sys.Diff = [0.01 0.01 10]*a;
Sys.DiffFrame = [0 53 45]*pi/180;
R = erot(Sys.DiffFrame);
Sys.Diffdiag = diag(Sys.Diff);
DiffM = R*Sys.Diffdiag*R.';
Sys.lw = [0.01,.065];
Exp.mwFreq = 8.90680900;
Exp.CenterSweep = [317.9120, 30];
Opt.Verbosity = 1;
[B2,spc2] = chili(Sys,Exp,Opt);
plot(B2,spc2)
It generated the following output:
Code: Select all
-- slow motion regime simulation ----------------------------------
field sweep, mw frequency 8.906809 GHz
field range (mT): min 302.912, max 332.912, center 317.912, width 30
harmonic 1, perpendicular mode
No ordering potential given, skipping powder simulation.
Solver: Lanczos tridiagonalization, left-to-right continued fraction evaluation
using S=1/2 Liouvillian code
Ordering potential: absent
Setting up basis set...
spatial basis: Leven max 14, Lodd max 7, Kmax 6, Mmax 2, deltaK 1, jKmin -1
spin basis: pSmin +0, pImax 2
M-p symmetry: 0
basis size: 15120
Computing starting vector...
vector size: 15120x1
non-zero elements: 3/15120 (0.02%)
maxabs 0.57735, norm 1
Computing Liouvillian matrix...
size: 15120x15120, maxabs: 1
non-zero elements: 2054856/228614400 (0.90%)
Computing spectrum...
converged to within 1e-06 at iteration 230/15120
Convoluting with Gaussian (FWHM 0.01 mT)...
-------------------------------------------------------------------
and the following plot:
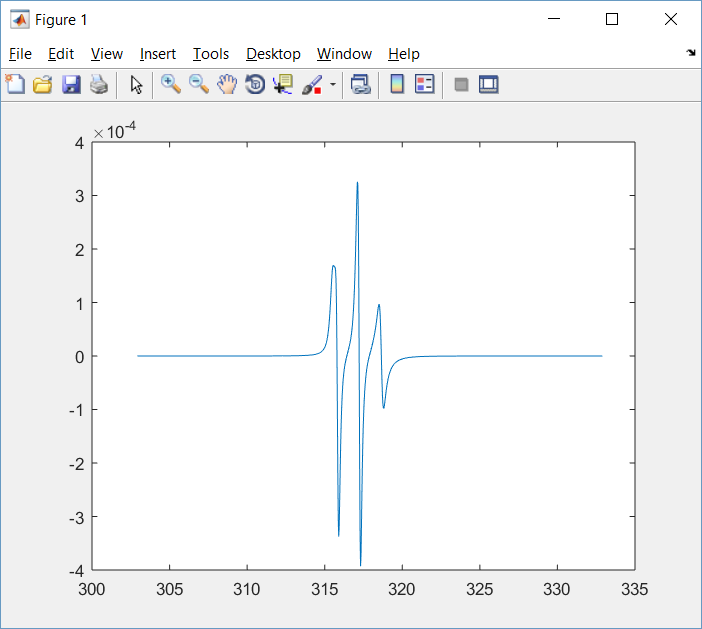
- nitroxide_chili.png (26.11 KiB) Viewed 6805 times
I ran this with ML 2016b and ES 5.1.6. Can you reproduce this on one of your computers? If not, I have no idea what could be wrong.